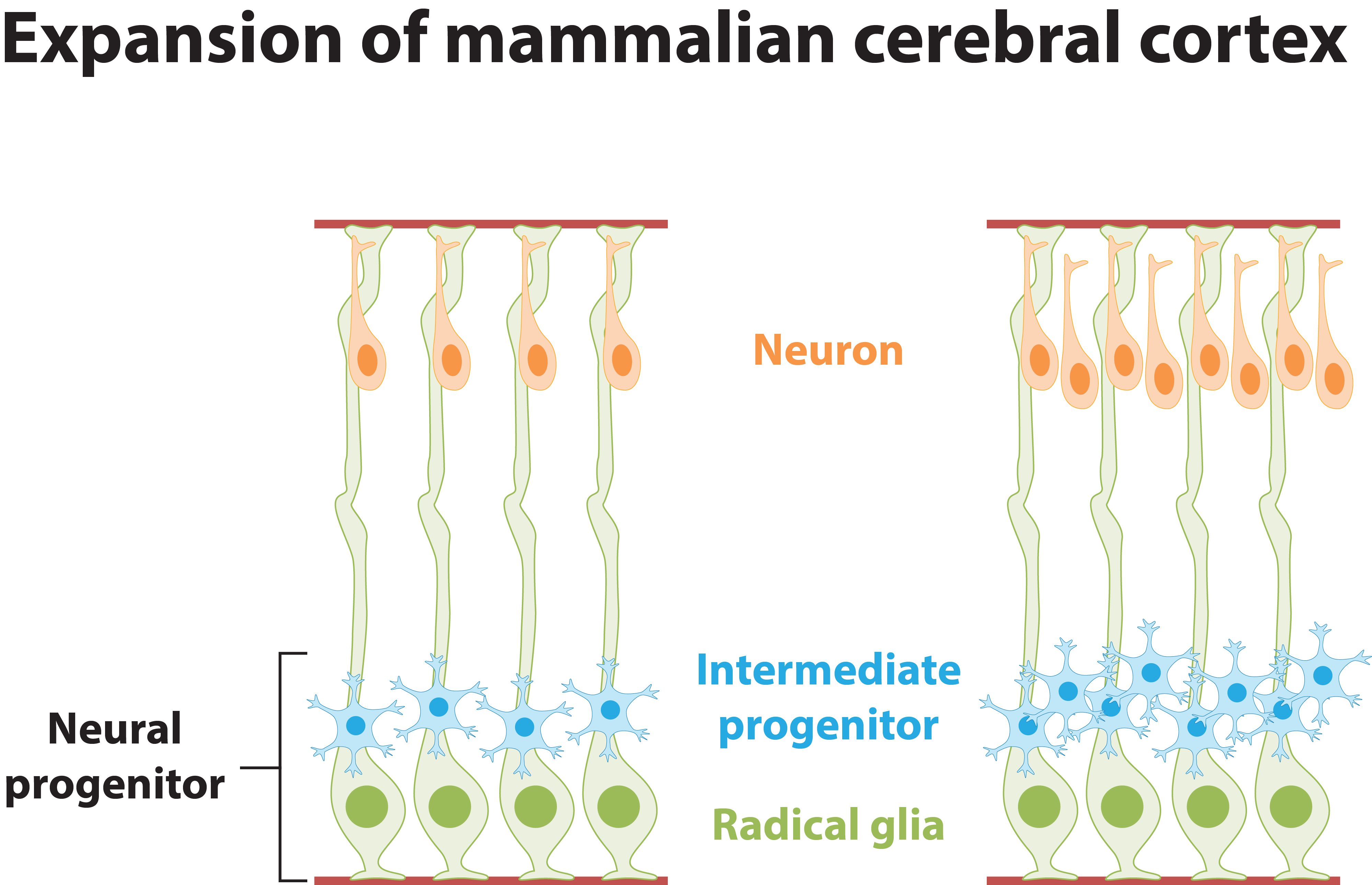
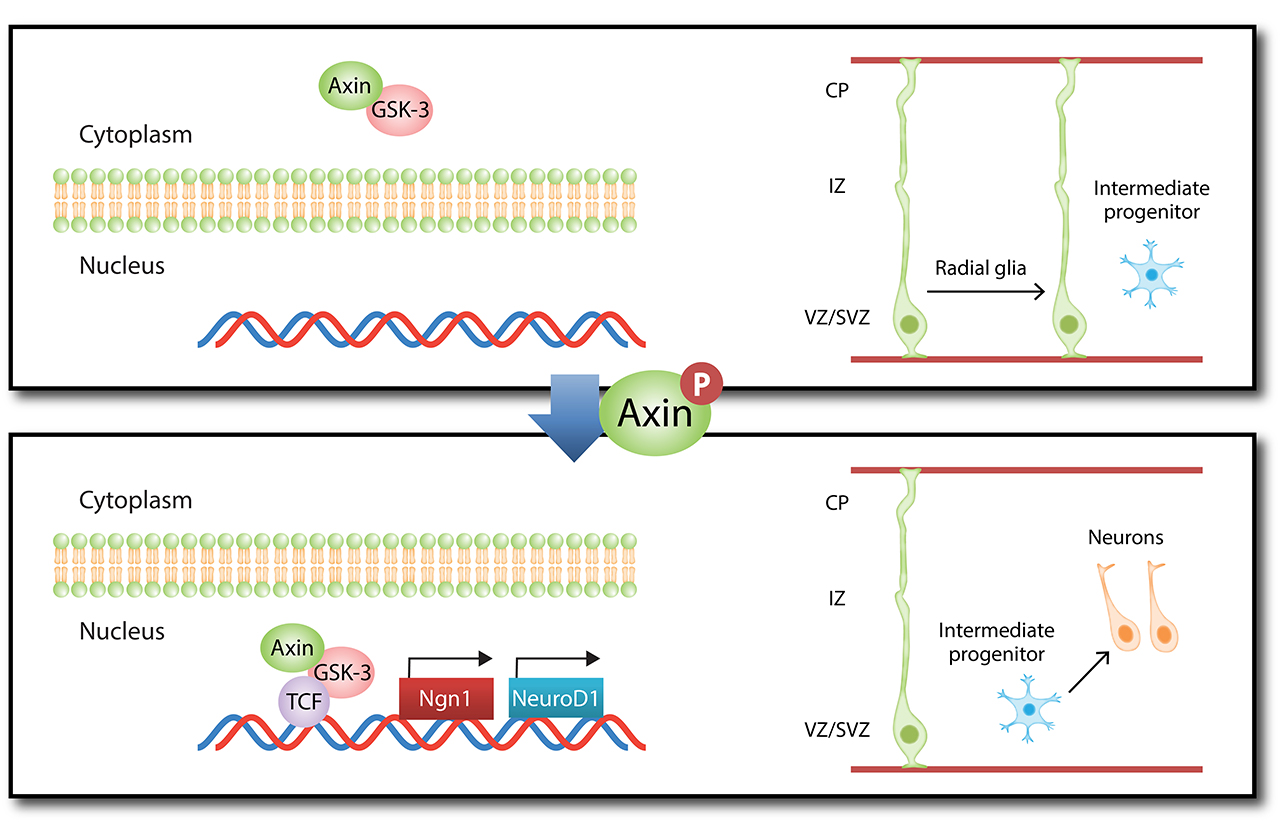
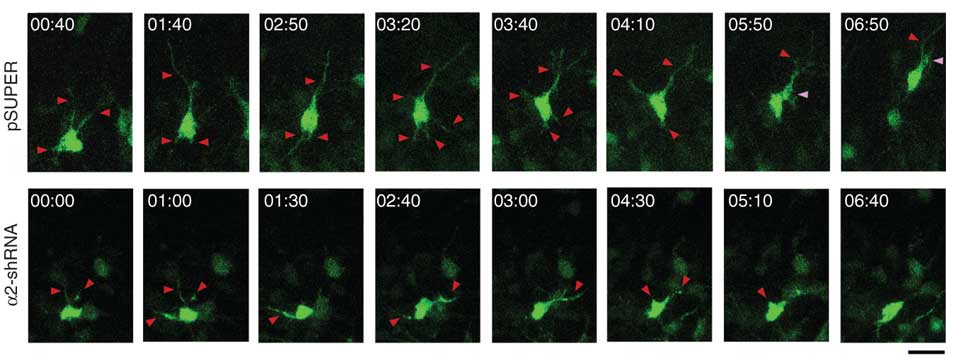
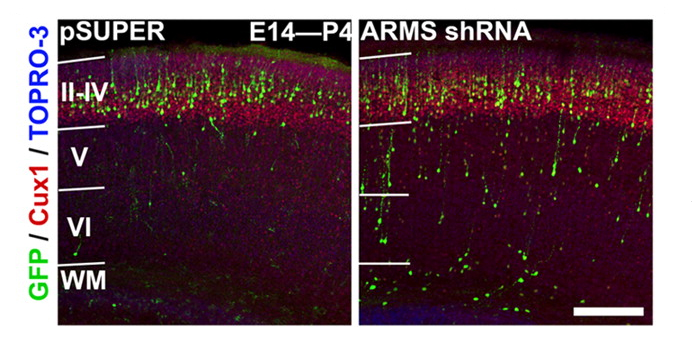
The mammalian cerebral cortex comprises six layers of highly laminated neurons generated through coordinated neurogenesis and migration in the embryonic stage. The mammalian cerebral cortex arises during forebrain formation from a sheet of proliferative cells in the dorsal telencephalon. Neural progenitors, also known as radial glial cells, proliferate in the ventricular zone and differentiate, and newborn neurons subsequently exit the ventricular zone and migrate to their final position. Neurons then send their axons to innervate targets and extend their dendrites to receive and integrate information from presynaptic partners. Thus, the proper development and functioning of neural circuitry depend on multiple cellular events, including the balanced proliferation and differentiation of neural progenitor cells, correct neuron positioning, and axon and dendrite formation. These steps are controlled by various molecular signaling pathways. Any defect in these processes can impair neural functions, which are associated with neurological disorders in human including lissencephaly (smooth brain), cortical heterotopias (double cortex), macrocephaly/microcephaly (large/small brain), and autism. Our laboratory investigates the specific signaling mechanisms that govern the different processes of neural circuit development.